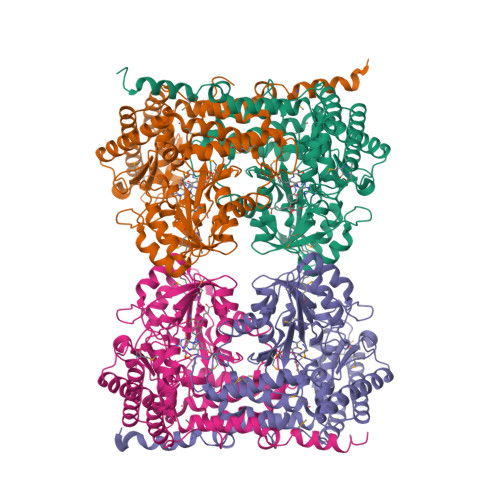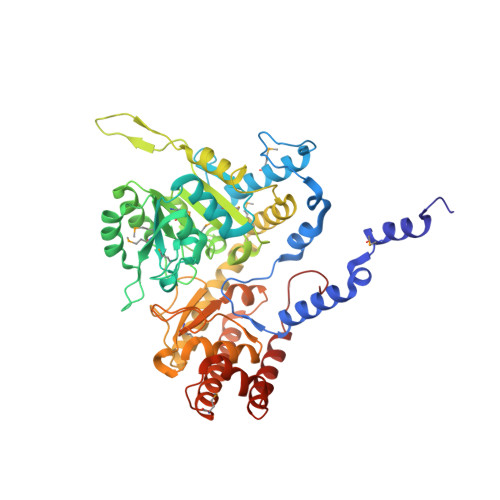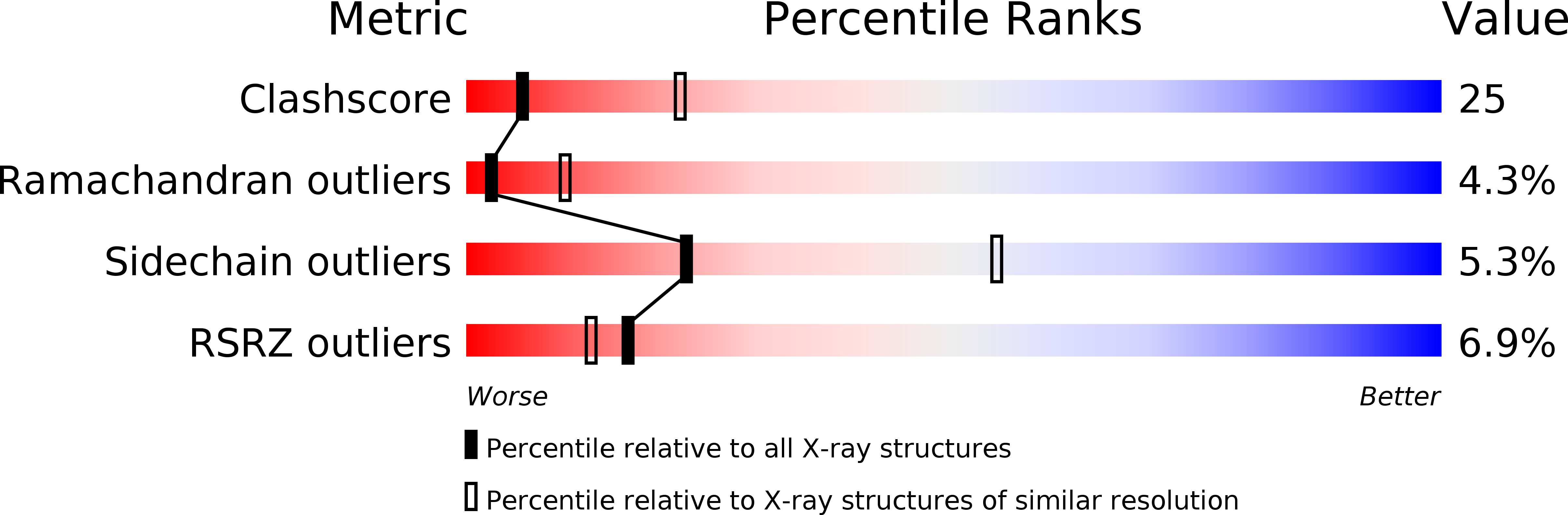
Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: evidence for asymmetric obligate dimers.
Szebenyi, D.M., Liu, X., Kriksunov, I.A., Stover, P.J., Thiel, D.J.(2000) Biochemistry 39: 13313-13323
- PubMed: 11063567
- DOI: https://doi.org/10.1021/bi000635a
- Primary Citation of Related Structures:
1EJI - PubMed Abstract:
Serine hydroxymethyltransferase (SHMT) is a pyridoxal phosphate-dependent enzyme that catalyzes the reversible conversion of serine and tetrahydrofolate to glycine and methylenetetrahydrofolate. This reaction generates single carbon units for purine, thymidine, and methionine biosynthesis. The enzyme is a homotetramer comprising two obligate dimers and four pyridoxal phosphate-bound active sites. The mammalian enzyme is present in cells in both catalytically active and inactive forms. The inactive form is a ternary complex that results from the binding of glycine and 5-formyltetrahydrofolate polyglutamate, a slow tight-binding inhibitor. The crystal structure of a close analogue of the inactive form of murine cytoplasmic SHMT (cSHMT), lacking only the polyglutamate tail of the inhibitor, has been determined to 2.9 A resolution. This first structure of a ligand-bound mammalian SHMT allows identification of amino acid residues involved in substrate binding and catalysis. It also reveals that the two obligate dimers making up a tetramer are not equivalent; one can be described as "tight-binding" and the other as "loose-binding" for folate. Both active sites of the tight-binding dimer are occupied by 5-formyltetrahydrofolate (5-formylTHF), whose N5-formyl carbon is within 4 A of the glycine alpha-carbon of the glycine-pyridoxal phosphate complex; the complex appears to be primarily in its quinonoid form. In the loose-binding dimer, 5-formylTHF is present in only one of the active sites, and its N5-formyl carbon is 5 A from the glycine alpha-carbon. The pyridoxal phosphates appear to be primarily present as geminal diamine complexes, with bonds to both glycine and the active site lysine. This structure suggests that only two of the four catalytic sites on SHMT are catalytically competent and that the cSHMT-glycine-5-formylTHF ternary complex is an intermediate state analogue of the catalytic complex associated with serine and glycine interconversion.
Organizational Affiliation:
Department of Molecular Biology and Genetics and Division of Nutritional Sciences, Cornell University, Ithaca, New York 14853, USA.