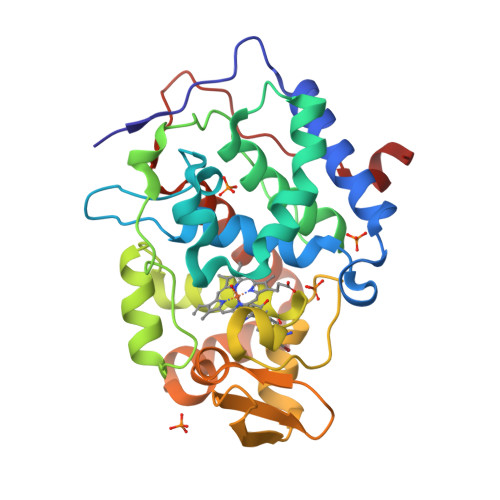
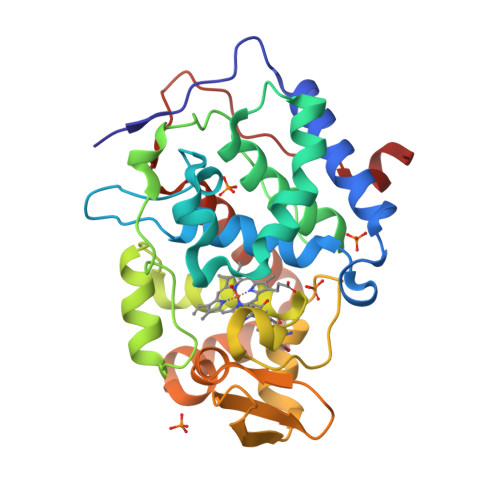
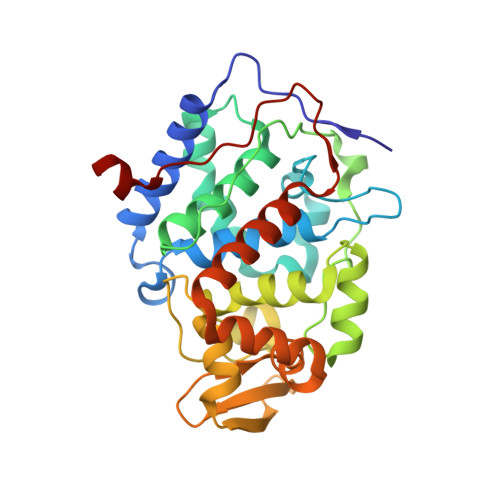
Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery.
Fischer, M., Coleman, R.G., Fraser, J.S., Shoichet, B.K.(2014) Nat Chem 6: 575-583
- PubMed: 24950326
- DOI: https://doi.org/10.1038/nchem.1954
- Primary Citation of Related Structures:
4NVA, 4NVB, 4NVC, 4NVD, 4NVE, 4NVF, 4NVG, 4NVH, 4NVI, 4NVJ, 4NVK, 4NVL, 4NVM, 4NVN, 4NVO, 4OQ7 - PubMed Abstract:
Proteins fluctuate between alternative conformations, which presents a challenge for ligand discovery because such flexibility is difficult to treat computationally owing to problems with conformational sampling and energy weighting. Here we describe a flexible docking method that samples and weights protein conformations using experimentally derived conformations as a guide. The crystallographically refined occupancies of these conformations, which are observable in an apo receptor structure, define energy penalties for docking. In a large prospective library screen, we identified new ligands that target specific receptor conformations of a cavity in cytochrome c peroxidase, and we confirm both ligand pose and associated receptor conformation predictions by crystallography. The inclusion of receptor flexibility led to ligands with new chemotypes and physical properties. By exploiting experimental measures of loop and side-chain flexibility, this method can be extended to the discovery of new ligands for hundreds of targets in the Protein Data Bank for which similar experimental information is available.
Organizational Affiliation:
1] Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, California 94158, USA [2] Faculty of Pharmacy, Donnelly Center, University of Toronto, 160 College St, Toronto, Ontario M5S 3E1 Canada [3].