



Structural insights into inhibition of lipid I production in bacterial cell wall synthesis.
Chung, B.C., Mashalidis, E.H., Tanino, T., Kim, M., Matsuda, A., Hong, J., Ichikawa, S., Lee, S.Y.(2016) Nature 533: 557-560
- PubMed: 27088606
- DOI: https://doi.org/10.1038/nature17636
- Primary Citation of Related Structures:
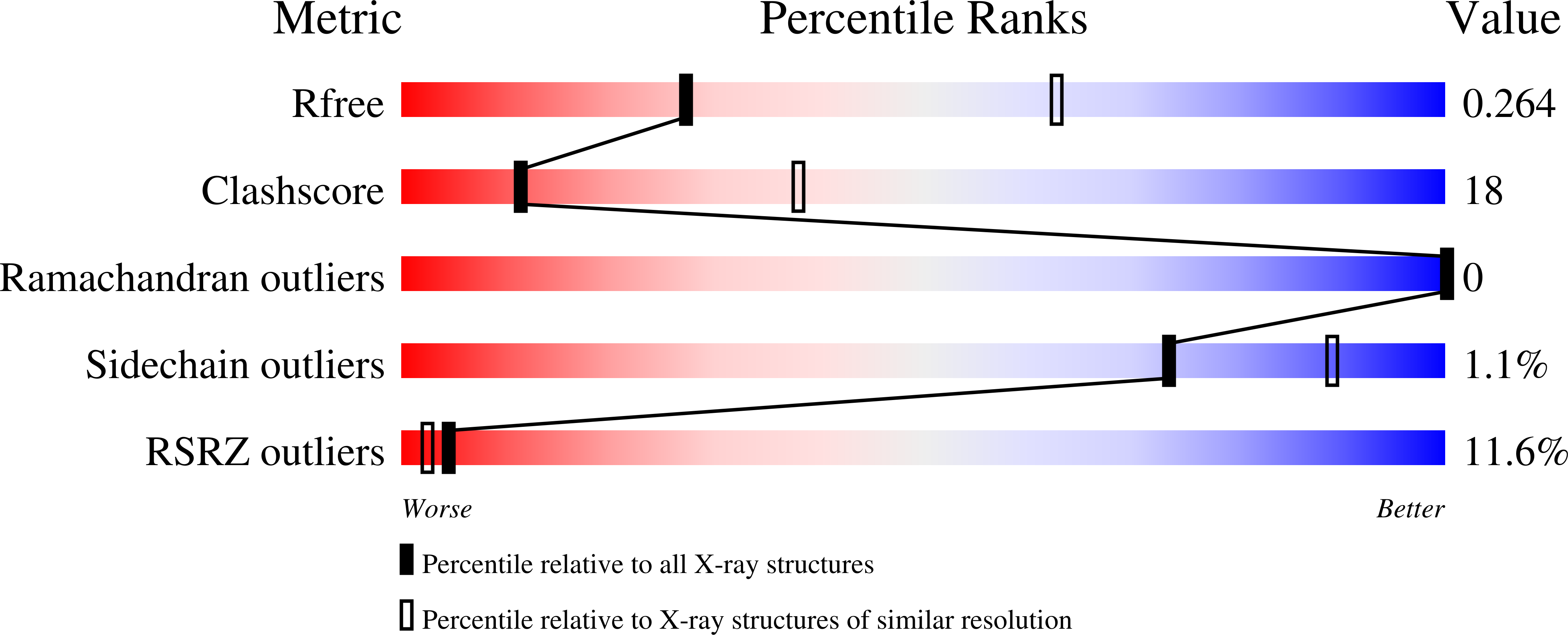
5CKR - PubMed Abstract:
Antibiotic-resistant bacterial infection is a serious threat to public health. Peptidoglycan biosynthesis is a well-established target for antibiotic development. MraY (phospho-MurNAc-pentapeptide translocase) catalyses the first and an essential membrane step of peptidoglycan biosynthesis. It is considered a very promising target for the development of new antibiotics, as many naturally occurring nucleoside inhibitors with antibacterial activity target this enzyme. However, antibiotics targeting MraY have not been developed for clinical use, mainly owing to a lack of structural insight into inhibition of this enzyme. Here we present the crystal structure of MraY from Aquifex aeolicus (MraYAA) in complex with its naturally occurring inhibitor, muraymycin D2 (MD2). We show that after binding MD2, MraYAA undergoes remarkably large conformational rearrangements near the active site, which lead to the formation of a nucleoside-binding pocket and a peptide-binding site. MD2 binds the nucleoside-binding pocket like a two-pronged plug inserting into a socket. Further interactions it makes in the adjacent peptide-binding site anchor MD2 to and enhance its affinity for MraYAA. Surprisingly, MD2 does not interact with three acidic residues or the Mg(2+) cofactor required for catalysis, suggesting that MD2 binds to MraYAA in a manner that overlaps with, but is distinct from, its natural substrate, UDP-MurNAc-pentapeptide. We have determined the principles of MD2 binding to MraYAA, including how it avoids the need for pyrophosphate and sugar moieties, which are essential features for substrate binding. The conformational plasticity of MraY could be the reason that it is the target of many structurally distinct inhibitors. These findings can inform the design of new inhibitors targeting MraY as well as its paralogues, WecA and TarO.
Organizational Affiliation:
Department of Biochemistry, Duke University Medical Center, 303 Research Drive, Durham, North Carolina, 27710, USA.






