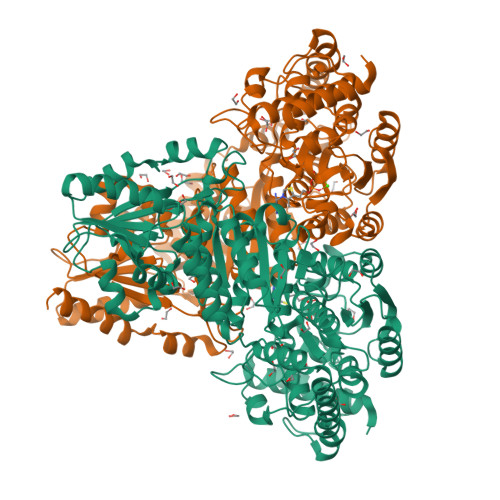
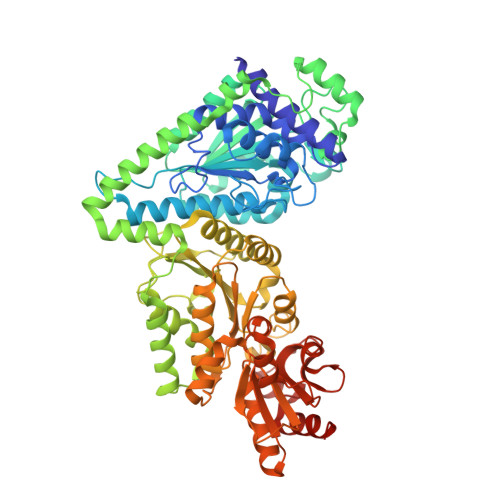
Structural Analysis of an Evolved Transketolase Reveals Divergent Binding Modes.
Affaticati, P.E., Dai, S.B., Payongsri, P., Hailes, H.C., Tittmann, K., Dalby, P.A.(2016) Sci Rep 6: 35716-35716
- PubMed: 27767080
- DOI: https://doi.org/10.1038/srep35716
- Primary Citation of Related Structures:
5HHT - PubMed Abstract:
The S385Y/D469T/R520Q variant of E. coli transketolase was evolved previously with three successive smart libraries, each guided by different structural, bioinformatical or computational methods. Substrate-walking progressively shifted the target acceptor substrate from phosphorylated aldehydes, towards a non-phosphorylated polar aldehyde, a non-polar aliphatic aldehyde, and finally a non-polar aromatic aldehyde. Kinetic evaluations on three benzaldehyde derivatives, suggested that their active-site binding was differentially sensitive to the S385Y mutation. Docking into mutants generated in silico from the wild-type crystal structure was not wholly satisfactory, as errors accumulated with successive mutations, and hampered further smart-library designs. Here we report the crystal structure of the S385Y/D469T/R520Q variant, and molecular docking of three substrates. This now supports our original hypothesis that directed-evolution had generated an evolutionary intermediate with divergent binding modes for the three aromatic aldehydes tested. The new active site contained two binding pockets supporting π-π stacking interactions, sterically separated by the D469T mutation. While 3-formylbenzoic acid (3-FBA) preferred one pocket, and 4-FBA the other, the less well-accepted substrate 3-hydroxybenzaldehyde (3-HBA) was caught in limbo with equal preference for the two pockets. This work highlights the value of obtaining crystal structures of evolved enzyme variants, for continued and reliable use of smart library strategies.
Organizational Affiliation:
Department of Biochemical Engineering, Gordon Street, University College London, WC1H 0AH, UK.