Targeting a Large Active Site: Structure-Based Design of Nanomolar Inhibitors of Trypanosoma brucei Trypanothione Reductase.
De Gasparo, R., Halgas, O., Harangozo, D., Kaiser, M., Pai, E.F., Krauth-Siegel, R.L., Diederich, F.(2019) Chemistry 25: 11416-11421
- PubMed: 31407832
- DOI: https://doi.org/10.1002/chem.201901664
- Primary Citation of Related Structures:
6OEX, 6OEY, 6OEZ - PubMed Abstract:
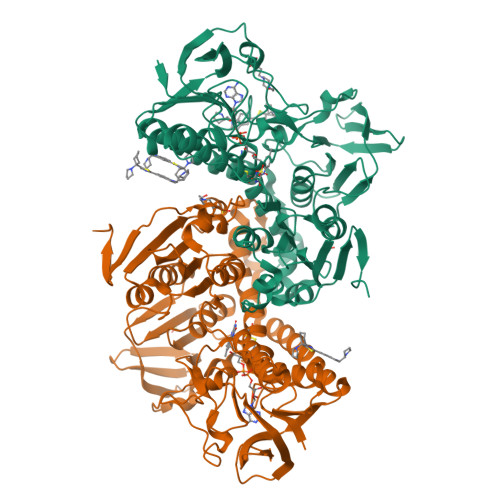
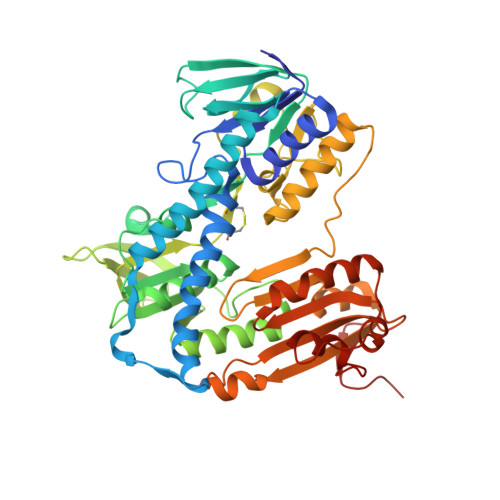
Trypanothione reductase (TR) plays a key role in the unique redox metabolism of trypanosomatids, the causative agents of human African trypanosomiasis (HAT), Chagas' disease, and leishmaniases. Introduction of a new, lean propargylic vector to a known class of TR inhibitors resulted in the strongest reported competitive inhibitor of Trypanosoma (T.) brucei TR, with an inhibition constant K i of 73 nm, which is fully selective against human glutathione reductase (hGR). The best ligands exhibited in vitro IC 50 values (half-maximal inhibitory concentration) against the HAT pathogen, T. brucei rhodesiense, in the mid-nanomolar range, reaching down to 50 nm. X-Ray co-crystal structures confirmed the binding mode of the ligands and revealed the presence of a HEPES buffer molecule in the large active site. Extension of the propargylic vector, guided by structure-based design, to replace the HEPES buffer molecule should give inhibitors with low nanomolar K i and IC 50 values for in vivo studies.
Organizational Affiliation:
Laboratorium für Organische Chemie, ETH Zurich, Vladimir-Prelog-Weg 3, 8093, Zurich, Switzerland.