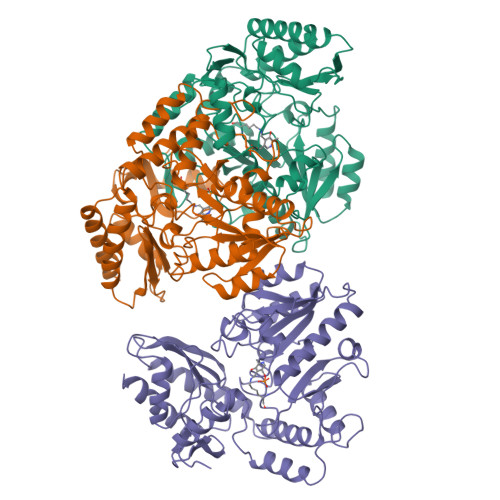
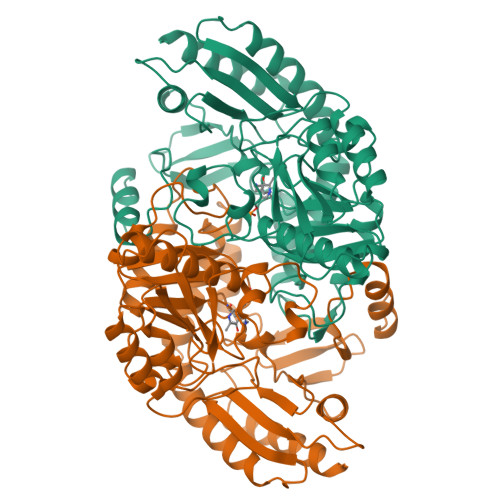
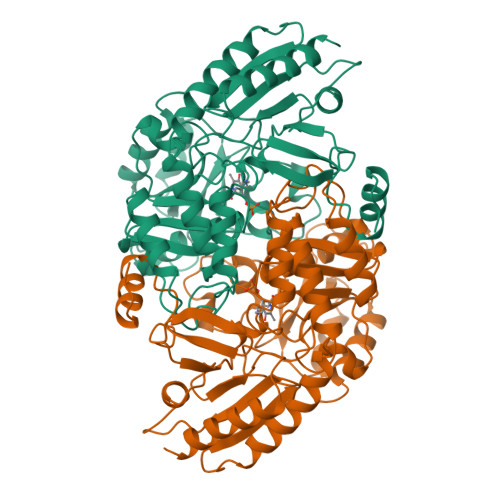
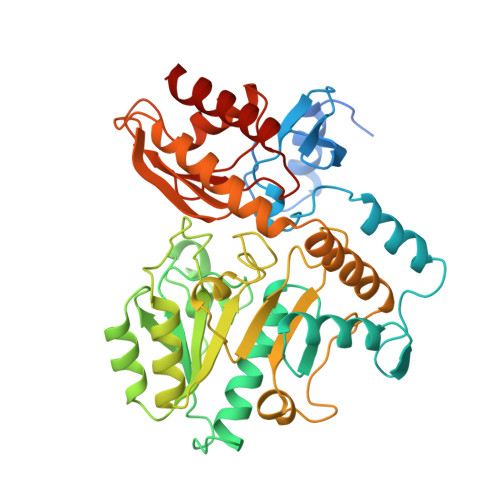
Crystal structure of human recombinant ornithine aminotransferase.
Shen, B.W., Hennig, M., Hohenester, E., Jansonius, J.N., Schirmer, T.(1998) J Mol Biol 277: 81-102
- PubMed: 9514741
- DOI: https://doi.org/10.1006/jmbi.1997.1583
- Primary Citation of Related Structures:
1OAT - PubMed Abstract:
Ornithine aminotransferase (OAT), a pyridoxal-5'-phosphate dependent enzyme, catalyses the transfer of the delta-amino group of L-ornithine to 2-oxoglutarate, producing L-glutamate-gamma-semialdehyde, which spontaneously cyclizes to pyrroline-5-carboxylate, and L-glutamate. The crystal structure determination of human recombinant OAT is described in this paper. As a first step, the structure was determined at low resolution (6 A) by molecular replacement using the refined structure of dialkylglycine decarboxylase as a search model. Crystallographic phases were then refined and extended in a step-wise fashion to 2.5 A by cyclic averaging of the electron density corresponding to the three monomers within the asymmetric unit. Interpretation of the resulting map was straightforward and refinement of the model resulted in an R-factor of 17.1% (Rfree=24.3%). The success of the procedure demonstrates the power of real-space molecular averaging even with only threefold redundancy. The alpha6-hexameric molecule is a trimer of intimate dimers with a monomer-monomer interface of 5500 A2 per subunit. The three dimers are related by an approximate 3-fold screw axis with a translational component of 18 A. The monomer fold is that of a typical representative of subgroup 2 aminotransferases and very similar to those described for dialkylglycine decarboxylase from Pseudomonas cepacia and glutamate-1-semialdehyde aminomutase from Synechococcus. It consists of a large domain that contributes most to the subunit interface, a C-terminal small domain most distant to the 2-fold axis and an N-terminal region that contains a helix, a loop and a three stranded beta-meander embracing a protrusion in the large domain of the second subunit of the dimer. The large domain contains the characteristic central seven-stranded beta-sheet (agfedbc) covered by eight helices in a typical alpha/beta fold. The cofactor pyridoxal-5'-phosphate is bound through a Schiff base to Lys292, located in the loop between strands f and g. The C-terminal domain includes a four-stranded antiparallel beta-sheet in contact with the large domain and three further helices at the far end of the subunit. The active sites of the dimer lie, about 25 A apart, at the subunit and domain interfaces. The conical entrances are on opposite sides of the dimer. In the active site, R180, E235 and R413 are probable substrate binding residues. Structure-based sequence comparisons with related transaminases in this work support that view. In patients suffering from gyrate atrophy, a recessive hereditary genetic disorder that can cause blindness in humans, ornithine aminotransferase activity is lacking. A large number of frameshift and point mutations in the ornithine aminotransferase gene have been identified in such patients. Possible effects of the various point mutations on the structural stability or the catalytic competence of the enzyme are discussed in light of the three-dimensional structure.
Organizational Affiliation:
Department of Structural Biology, Biozentrum, Basel, CH-4056, Switzerland.