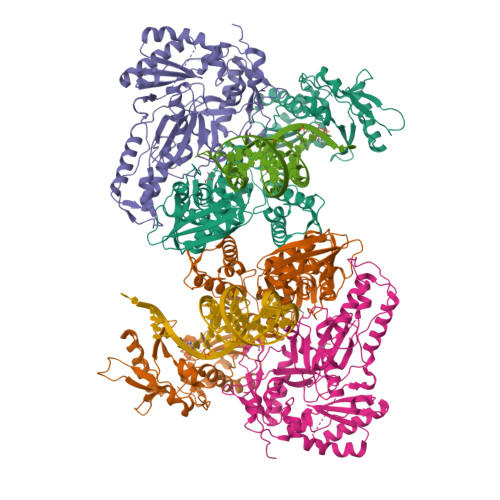
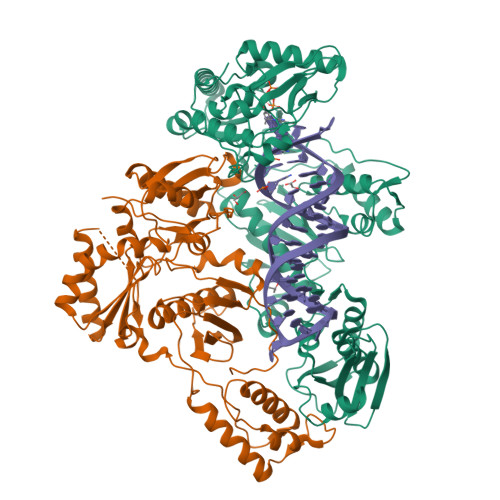
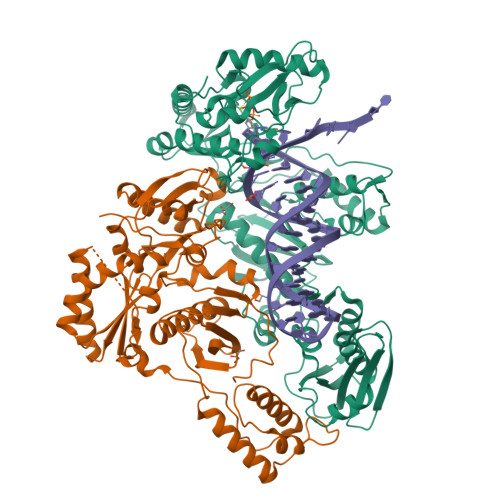
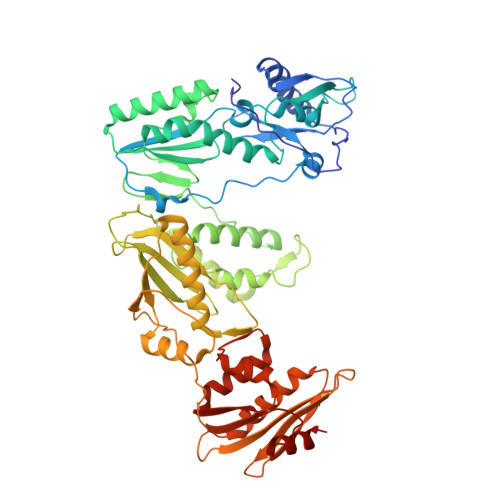
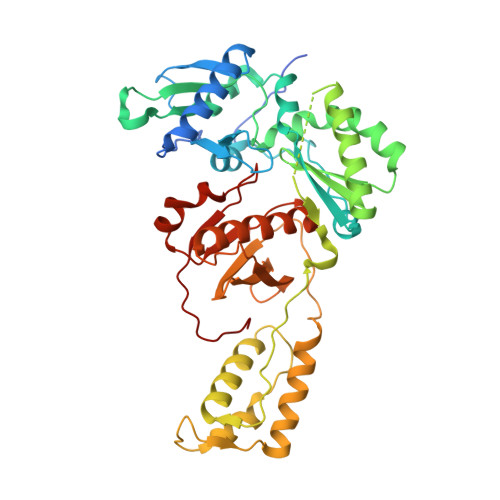
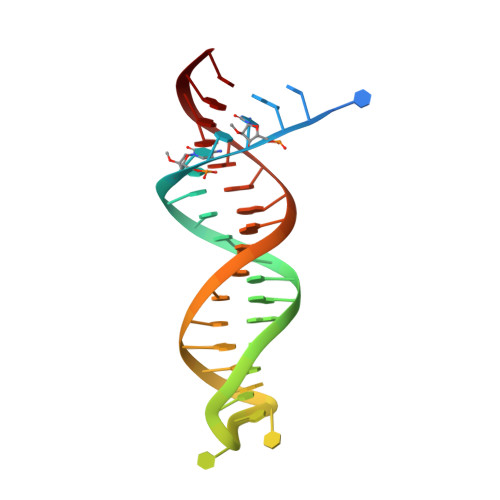
Deviated binding of anti-HBV nucleoside analog E-CFCP-TP to the reverse transcriptase active site attenuates the effect of drug-resistant mutations.
Yasutake, Y., Hattori, S.I., Kumamoto, H., Tamura, N., Maeda, K., Mitsuya, H.(2024) Sci Rep 14: 15742-15742
- PubMed: 38977798
- DOI: https://doi.org/10.1038/s41598-024-66505-z
- Primary Citation of Related Structures:
8X1Z, 8X20, 8X21, 8X22 - PubMed Abstract:
While certain human hepatitis B virus-targeting nucleoside analogs (NAs) serve as crucial anti-HBV drugs, HBV yet remains to be a major global health threat. E-CFCP is a 4'-modified and fluoromethylenated NA that exhibits potent antiviral activity against both wild-type and drug-resistant HBVs but less potent against human immunodeficiency virus type-1 (HIV-1). Here, we show that HIV-1 with HBV-associated amino acid substitutions introduced into the RT's dNTP-binding site (N-site) is highly susceptible to E-CFCP. We determined the X-ray structures of HBV-associated HIV-1 RT mutants complexed with DNA:E-CFCP-triphosphate (E-CFCP-TP). The structures revealed that exocyclic fluoromethylene pushes the Met184 sidechain backward, and the resultant enlarged hydrophobic pocket accommodates both the fluoromethylene and 4'-cyano moiety of E-CFCP. Structural comparison with the DNA:dGTP/entecavir-triphosphate complex also indicated that the cyclopentene moiety of the bound E-CFCP-TP is slightly skewed and deviated. This positioning partly corresponds to that of the bound dNTP observed in the HIV-1 RT mutant with drug-resistant mutations F160M/M184V, resulting in the attenuation of the structural effects of F160M/M184V substitutions. These results expand our knowledge of the interactions between NAs and the RT N-site and should help further design antiviral NAs against both HIV-1 and HBV.
Organizational Affiliation:
Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Sapporo, 062-8517, Japan. y-yasutake@aist.go.jp.