Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential.
Prade, L., Engh, R.A., Girod, A., Kinzel, V., Huber, R., Bossemeyer, D.(1997) Structure 5: 1627-1637
- PubMed: 9438863
- DOI: https://doi.org/10.1016/s0969-2126(97)00310-9
- Primary Citation of Related Structures:
1STC - PubMed Abstract:
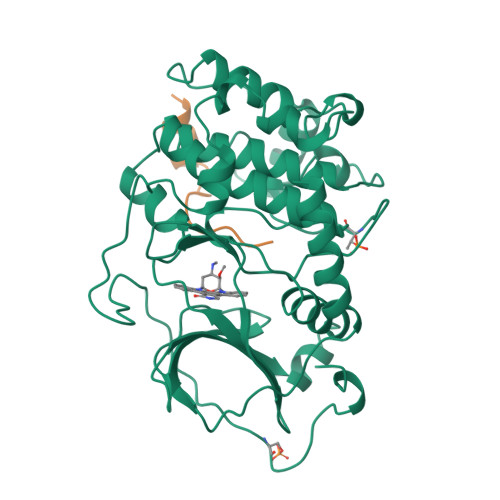
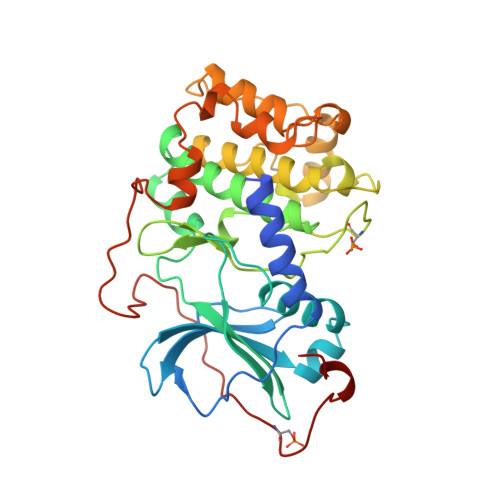
Staurosporine inhibits most protein kinases at low nanomolar concentrations. As most tyrosine kinases, along with many serine/threonine kinases, are either proto oncoproteins or are involved in oncogenic signaling, the development of protein kinase inhibitors is a primary goal of cancer research. Staurosporine and many of its derivatives have significant biological effects, and are being tested as anticancer drugs. To understand in atomic detail the mode of inhibition and the parameters of high-affinity binding of staurosporine to protein kinases, the molecule was cocrystallized with the catalytic subunit of cAMP-dependent protein kinase. The crystal structure of the protein kinase catalytic subunit with staurosporine bound to the adenosine pocket shows considerable induced-fit rearrangement of the enzyme and a unique open conformation. The inhibitor mimics several aspects of adenosine binding, including both polar and nonpolar interactions with enzyme residues, and induces conformational changes of neighboring enzyme residues. The results explain the high inhibitory potency of staurosporine, and also illustrate the flexibility of the protein kinase active site. The structure, therefore, is not only useful for the design of improved anticancer therapeutics and signaling drugs, but also provides a deeper understanding of the conformational flexibility of the protein kinase.
Organizational Affiliation:
Abteilung Strukturforschung Max-Planck-Institut für Biochemie, Martinsried, Germany.